
Generative Genomics: Predicting Future Experiments with GEM-1
GEM-1 is a generative AI system that predicts gene expression experimental results from experimental descriptions.

Gene expression measurement via RNA-seq is foundational to modern biomedical research. Yet, every experiment remains constrained by fundamental biological limits. Cells grow at their own pace. Diseases develop over time. Clinical trials depend on patient recruitment and regulatory processes. Inherent biological complexity makes predicting results, or even asking the most fruitful questions, difficult.
What if we could computationally simulate the results of experiments we haven’t yet run? That is exactly the premise of, GEM-1 (Generate Expression Model-1), the first model we released.
Our preprint1 gives you all the details, but we wanted to provide a more casual overview. Read on…
GEM-1 is a deep latent variable model. It takes experimental metadata as input and generates predicted gene expression profiles as output. The system effectively mirrors the process of designing and conducting an RNA-seq experiment: you describe what you want to study, and GEM-1 predicts what you’d observe.
This work represents a step toward generative genomics: moving beyond cataloging data to simulating the limiting steps of laboratory experimentation.
The challenge with gene expression modeling
Foundation models for single-cell gene expression data have made impressive progress on tasks like cell type annotation, batch correction, and perturbation prediction. But most models are trained on curated single-cell atlases with uniform processing. Therefore, they struggle with the heterogeneous, incompletely annotated data. For better or worse, those messy data constitute most real-world research.
We extend the idea behind these models with a new goal: a generative system that captures the full diversity of gene expression experiments. This includes basic research, preclinical studies, clinical trials, bulk and single-cell, healthy and diseased. We plan to predict experimental outcomes rather than embedding or classifying existing data.
Building a training set that reflects real research
To assemble a “real world” gene expression training dataset, the critical challenge wasn’t obtaining data. Rather, it was creating usable experimental descriptions from the fragmented, inconsistently annotated metadata that accompanies public submissions.
We built a metadata agent using LLMs to convert raw SRA metadata into harmonized descriptions with controlled vocabulary covering experimental parameters: cell type, tissue, disease, perturbation identity, dose, timing, and technical factors like library preparation and sequencing platform. The resulting dataset spans blood, brain, liver, and dozens of other tissues; cancers, immune disorders, and infectious diseases; over 18,000 distinct perturbations, including 650+ small molecules and 300+ pathogen infections.
We constructed our training dataset from 470,691 bulk RNA-seq samples across 24,715 datasets in the SRA, deposited through June 2024. For single-cell data, we trained on 41 million cells from CellxGene and scPerturb.org, representing 669 cell types across 259 tissues. (More on our training data coming soon.)
Architecture: Separable latent factors for interpretable generation
GEM-1 is a deep latent variable model that partitions experimental metadata into three independent sources: biological (sex, tissue, disease), technical (library prep, platform), and perturbational (compound identity, dose, time). Each source gets its own latent distribution, and these combine to parameterize the final distribution over gene expression.
This architecture enables different inference modes for different problems. For de novo generation, you provide metadata, and the model predicts expression. For reference conditioning, a novel feature we baked into GEM-1’s architecture, you can ground predictions in actual experimental data. It’s like fine-tuning without all the fuss. For example, predicting the effect of a gene knockdown in a specific cell line by conditioning on existing expression data from unperturbed cells of that line. The model also includes internal classifiers that can predict metadata from expression data, which is useful for filling in missing annotations or detecting labeling errors.
This separable latent structure is key. It lets us mix and match: sample biological and technical factors from a reference sample’s posterior while sampling perturbational factors from a prior conditioned on a novel treatment. This “conditional sampling” capability enables simulations such as predicting a drug’s effect on a specific patient sample.

Predicting future experiments
The real test is whether the model can predict experiments it’s never seen. We constructed holdout sets from data deposited after our training cutoff (July through September 2024), categorizing samples by novelty: previously observed contexts, novel genetic perturbations, novel chemical perturbations, or novel biological contexts.
We report results using gene Pearson correlation (rgene), a more stringent metric than sample-level correlation since it tests whether the model correctly ranks each gene’s expression across samples.
For fully synthetic predictions (metadata only), GEM-1 achieved rgene of 0.62-0.66 across novel perturbations. Reference conditioning substantially improved performance across all categories, pushing novel biological contexts to 0.75 and exceeding pseudoreplicate-level reproducibility across all holdout sets.

Benchmarking against lab reproducibility
How good is “good”? We established performance benchmarks by comparing replicate experiments (same study, matched metadata) and pseudoreplicates (different studies, matched metadata). These represent the ceiling of what any model could achieve: the inherent reproducibility of the lab experiments themselves.
GEM-1’s predictions matched pseudoreplicate-level performance in most settings without reference conditioning (or what we call “fully synthetic” mode). What’s more, with reference conditioning, GEM-1’s predictions exceeded pseudoreplicate reproducibility. The model correctly predicted over half of enriched gene sets in future experiments for novel perturbations, rising to 63-70% with reference conditioning.
Extension to single-cell data
GEM-1’s architecture handles single-cell data naturally. We evaluated on Tabula Sapiens v2 (additional organs and donors that were not seen in training for GEM-1 or models that we compared with). The model’s biological embeddings separate cell types comparably to scGPT, and it achieves competitive or better performance on cell type annotation using a linear probe framework, with macro F1 scores exceeding scGPT and SCimilarity across most tissues.
For generation, we compared predicted transcriptomes to replicate and pseudoreplicate benchmarks (same vs. different donors). GEM-1 predictions showed fairly consistent correlation across tissues, while lab reproducibility varied more widely.
Generating clinical cohorts
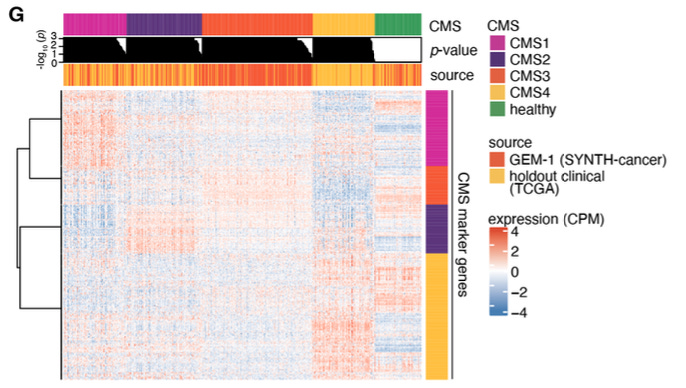
Perhaps the most striking application is generating data mimicking large clinical studies. We created three synthetic cohorts, excluding the corresponding real datasets from training:
SYNTH-TEx (5,300 samples, 30 tissues): AI-generated samples cluster with real GTEx data by tissue.
SYNTH-interferon (200 blood samples): Synthetic SLE samples show appropriate Type 1 interferon pathway upregulation.
SYNTH-cancer (10,523 samples, 27 cancer types): Samples conform to molecular subtyping schemes and show realistic expression of therapeutic targets like EGFR.
Limitations and future directions
These results are promising, but there are limitations. We focused on human data only, though the model architecture is applicable to other species. Our metadata schema, while comprehensive, doesn’t capture every experimental feature. But performance should improve as inputs become more fully specified. We also didn’t include extensive perturbational data (e.g., Perturb-seq data), which presents unique challenges around technical artifacts and appropriate evaluation metrics that warrant separate treatment.
The model’s ability to generalize to novel perturbations likely reflects not just our architecture, but the quality of pretrained embeddings we used to represent compounds and genes. As those embeddings improve, so should GEM-1’s extrapolation capability.
Toward computational simulation of biology
GEM-1 demonstrates that generative AI can predict gene expression experiments from experimental descriptions, including for perturbations never seen during training. The approach explicitly models the structure of biological experimentation by separating biological, technical, and perturbational factors. Thereby providing an interpretable framework for flexible in silico experimentation.
The ability to computationally simulate experiments before running them opens possibilities ranging from prioritizing which perturbations to test in the lab to generating synthetic cohort data for translational studies. GEM-1 represents an early step in that direction.
Learn More
You can explore the paper, the code, and our AI-generated datasets at the links below.
Read the Preprint: https://doi.org/10.1101/2025.09.08.674753
Explore the Datasets: https://app.synthesize.bio/datasets
The GEM-1 paper was co-authored by: Gregory Koytiger, Alice M. Walsh, Vaishali Marar, Kayla A. Johnson, Max Highsmith, Alexander R. Abbas, Andrew Stirn, Ariel R. Brumbaugh, Alex David, Darren Hui, Jeffrey M. Kahn, Sheng-Yong Niu, Liza J. Ray, Candace Savonen, Stein Setvik, Jeffrey T. Leek, and Robert K. Bradley
This is terrific work.
- Would you mind talking a bit more about the novel-cell-type demo? If you use one-hot encoding of cell types, then how does GEM-1 achieve a positive r_{gene}?
- For genetic perturbations, do you condition on the targeted gene? https://ekernf01.github.io/target_gene_shenanigans
- Do you have any ablation studies that omit the perturbation latents, and did you ever calculate r_gene within each individual test-set study?
Thanks very much and good luck to you!