
Virtual human modeling, at conversational speed
Synthesize Bio's Generative Expression Model (GEM-1) works with Claude

TL;DR: GEM-1, Synthesize Bio’s gene-expression foundation model for virtual human modeling, is now in Claude. All you need is a question. From there, Claude runs the experiment against GEM-1, our model trained on the largest annotated transcriptomic corpus ever assembled. It’s not just a faster way to do the old work — questions that weren’t answerable at all before are now within reach of anyone with a hypothesis. Our deepest work is done in partnership with pharma teams on clinical development decision support, spanning trial design, patient stratification, and biomarker discovery. The connector in Claude is an entry point — where any scientist can start exploring GEM-1.

Three things used to gate translational biology:
A biobank with access to human tissues across diseases and conditions
A platform to generate molecular data from those samples
A bioinformatics team that can turn the results into biological insight.
Most of the time scientists work without one or more of these, or with just fragments of them shared across competing priorities.
Synthesize Bio puts all three inside a Claude chat. It enables any biologist already working in Claude to reach Synthesize’s models without installing software, learning a new platform, or leaving the conversation they’re already in. Synthesize Bio + Claude enables the fast, powerful feedback loop that our twenty-plus year pharma veteran always wished he had on drug development projects.
Claude can directly access our gene expression model now.
Our Generative Expression Model enables a virtual lab on Claude
Describe the biological context, the compound or genetic perturbation, and the comparison you want. Claude translates that into queries against Synthesize’s GEM-1 model to generate lab-quality transcriptomic data, then it runs the analysis on the GEM-1-generated data, and returns results with its interpretation. You iterate and ask follow-ups in the same Claude chat session, without rebuilding a pipeline each time.
The output is a differential expression analysis (which genes changed), a pathway enrichment view (which biological programs those genes belong to), and a dashboard-style visualization (you can even make it interactive!). No samples, no wet lab, no code. The work that used to take a team a quarter or more now takes an afternoon.
If you’d like to skip ahead and try it for yourself, here’s the documentation and a getting-started guide.
Three experiments, one afternoon
Alex Abbas, PhD spent twenty years as a computational biologist in big pharma before joining Synthesize Bio as Director of Computational Biology. We asked him to push the system (Synthesize Bio + Claude) to its limits. The tool rewards a clear hypothesis: the sharper the biological question, the more useful the answer.
What follows is his account of where the system performed powerfully and some current limitations across three experiments.
1. Tissue versus cell line: modeling TGF-β in TF-1 cells vs MDS bone marrow
Summary. Drugs that look promising in lab-grown blood cell lines often behave very differently in actual human bone marrow. I asked Claude + GEM-1 to compare a TGF-β/ALK5 inhibitor in two contexts: TF-1 cells (a common lab stand-in) and virtual human bone marrow tissue. The cell line showed essentially no response. The bone marrow showed 283 differentially expressed genes hitting exactly the pathways you’d predict; red blood cell production machinery and inflammatory signaling. The bone marrow arm normally isn’t possible at all outside a clinical trial; both ran in minutes.
Details: Tissue versus cell line
Question. Myelodysplastic syndrome (MDS) is a bone marrow disorder that interferes with the body’s ability to produce healthy blood cells, and in many cases progresses to acute myeloid leukemia. The disease can be driven by disruption to a single signaling pathway that helps regulate how red blood cells mature; when that pathway breaks down, the result is the ineffective red-blood-cell production that defines MDS. Several drugs in development target this pathway, including elritercept (licensed by Takeda from Keros). A core question for any such program: does the drug behave the same way in human bone marrow as it does in the lab-grown cells researchers usually use as stand-ins?
Conversation & context. I asked Claude to use Synthesize Bio’s GEM-1 to compare gene expression changes from SB-431542, a well-characterized inhibitor of the related TGF-β/ALK5 branch, in two contexts: TF-1, a commonly used blood cell line, and human bone marrow tissue. The cell-line arm would normally take a month and substantial wet-lab resources. The tissue arm isn’t practical outside virtual human modeling. Claude + GEM-1 let me run both experiments in minutes.
Results. TF-1 returned zero statistically significant differentially expressed genes. Bone marrow returned 283: 119 up, 164 down. In plain terms, the tissue analysis flagged exactly the kind of genes you’d expect if the drug were hitting red-blood-cell production machinery and inflammatory signaling in the marrow. The downregulated set included ALAS2 and SLC25A37 (erythroid heme synthesis and mitochondrial iron import) and MXI1, a negative regulator of MYC. The upregulated set included IL1RL1, the receptor for IL-33, implicated in inflammatory signaling in hematopoietic progenitors. Together, these point to coherent biological effects on disease-relevant pathways.
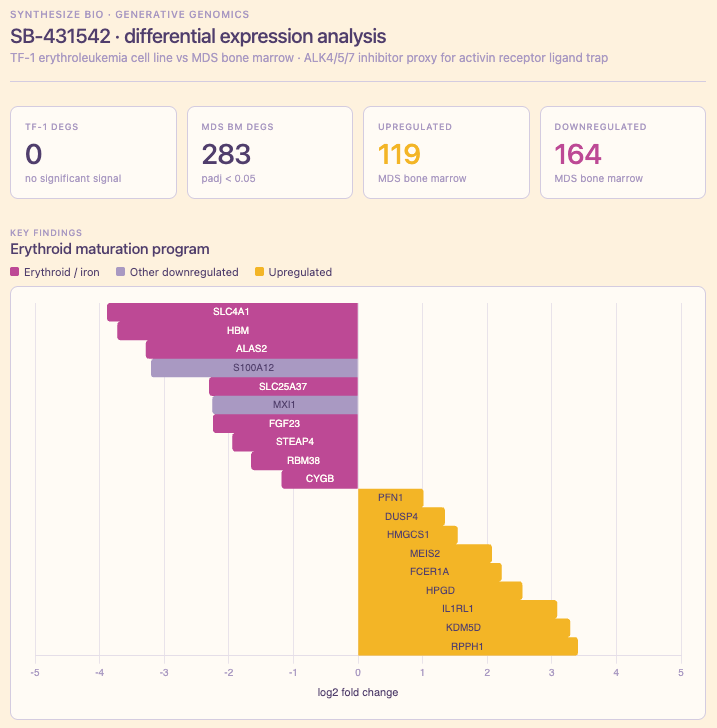
Interpretation. The absence of signal in TF-1 is as informative as the signal in bone marrow. It likely reflects a real limitation of that cell line as a model for this pathway in this disease… the kind of finding easy to miss when early work stays in cell culture, and one that can reshape experimental design well before a wet-lab program commits real resources.
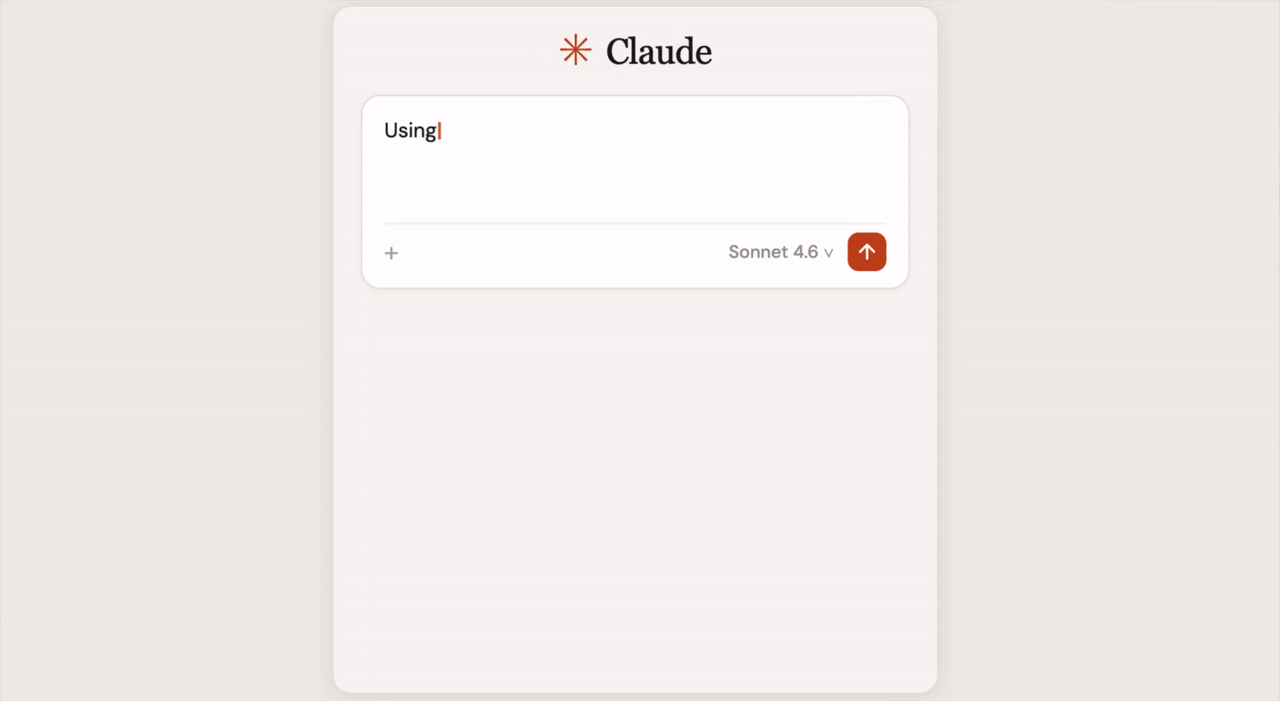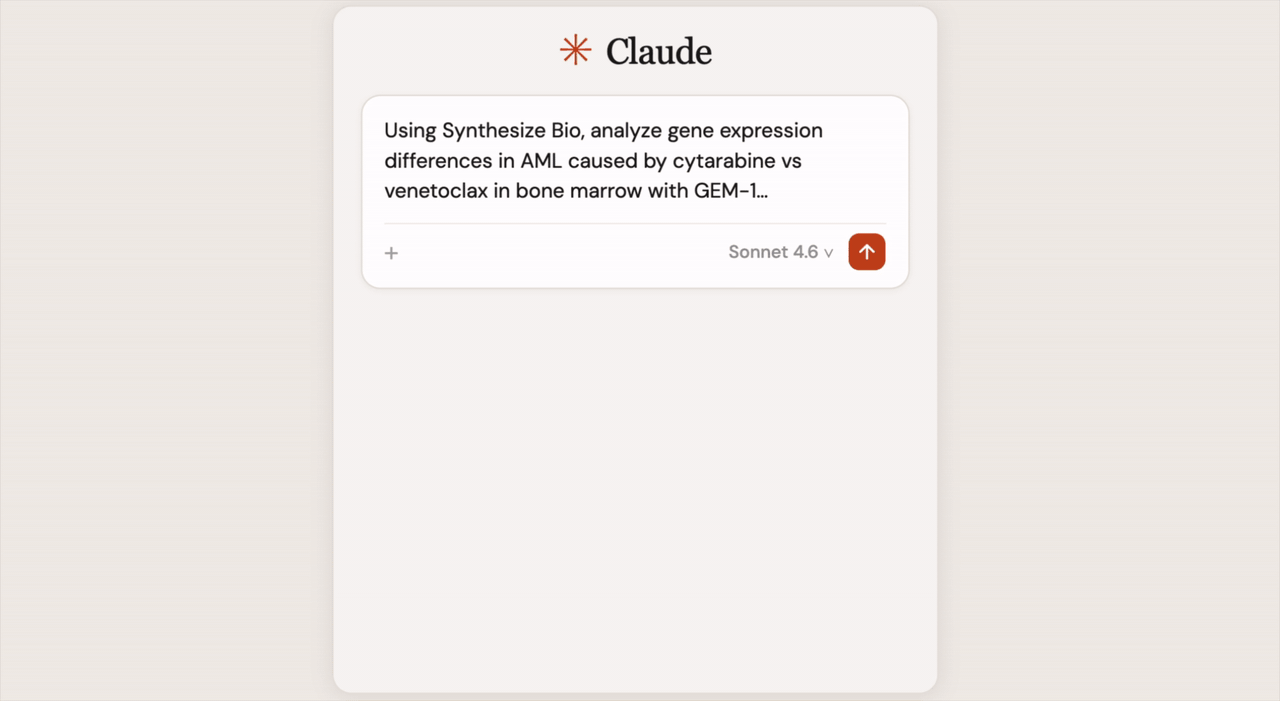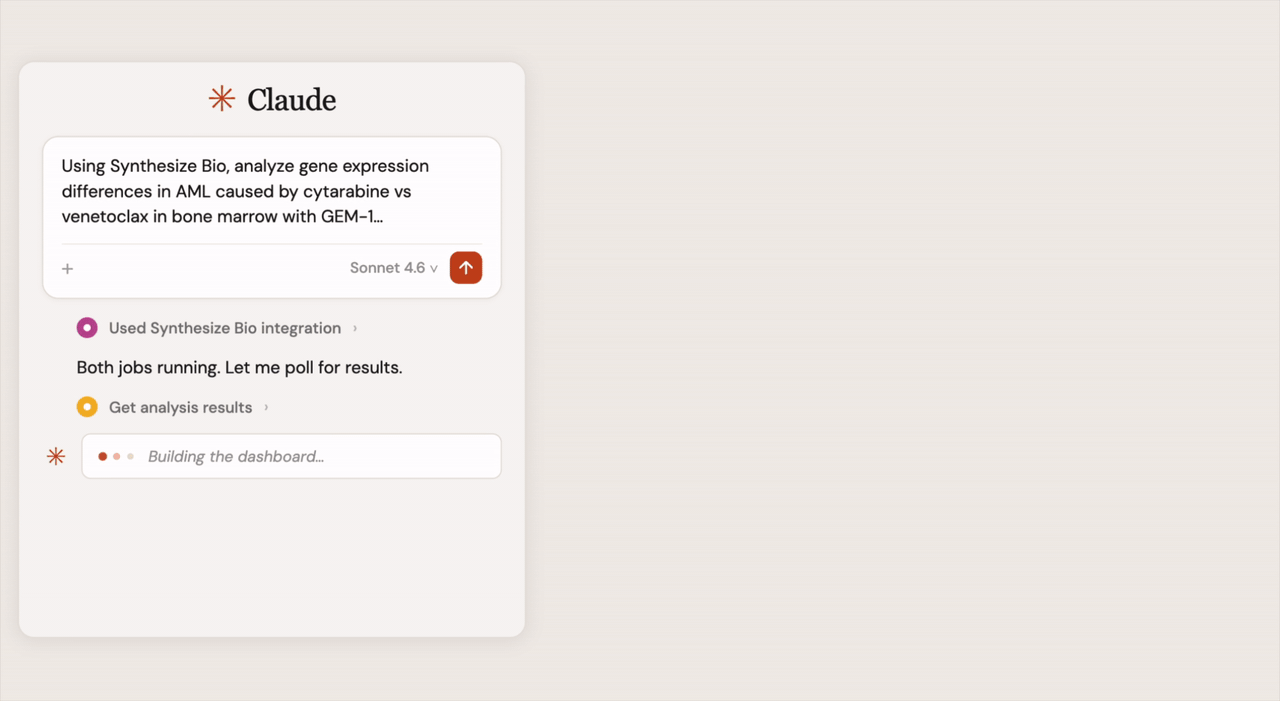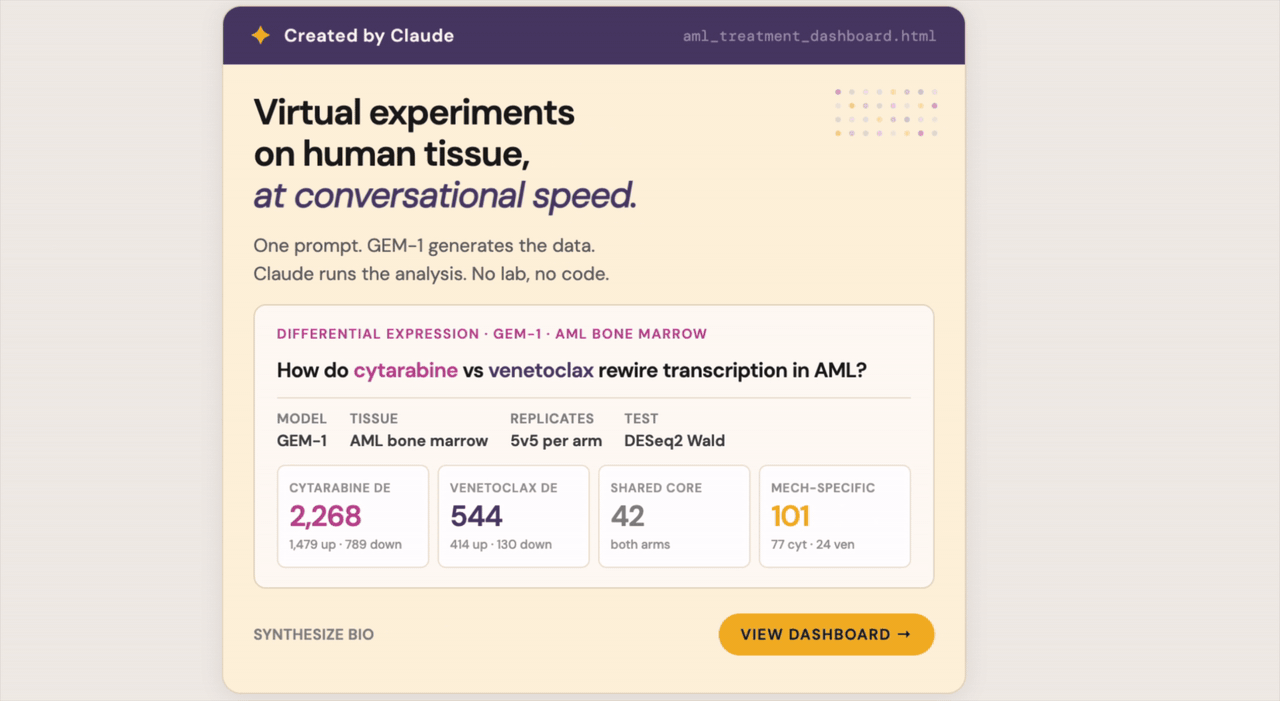
2. Comparative perturbation in AML: venetoclax versus cytarabine
Summary. Two AML drugs with very different mechanisms, an older broad-spectrum chemotherapy and a newer targeted agent, should leave very different fingerprints on which genes get turned on or off. I asked Claude + GEM-1 to put them side by side at the pathway level. Cytarabine touched the basic machinery cells use to copy and repair their DNA. Venetoclax stayed narrow, hitting the specific apoptosis-regulating protein it was designed for. The kind of comparison that normally takes a bioinformatician days to set up took just a few minutes of conversation.
Details: Comparative perturbation in AML
Question. Acute myeloid leukemia (AML) is one of the more therapeutically diverse blood cancers. Cytarabine is a decades-old chemotherapy that disrupts how cells copy their DNA, affecting many kinds of cells at once. Venetoclax is a newer, targeted drug that blocks a single protein cancer cells lean on to escape programmed cell death. The two leave very different fingerprints on which genes get turned on or off; understanding those differences shapes how doctors think about combining them, sequencing them, and predicting which patients will eventually stop responding. Can the system show what each drug is actually doing inside cells, side by side?
Conversation & context. Venetoclax is a well-known compound, but to build the comparison I needed a comparator that was represented in the training data. A practical approach: describe your desired experiment in general terms first and ask Claude to search Synthesize Bio for terms that will work. I started with “What are the major drugs used to treat AML besides venetoclax that the Synthesize Bio connector knows about?” and then launched the experiment with “Use the Synthesize Bio connector to analyze the differences in gene expression changes and pathway impacts in AML caused by treatment with cytarabine vs venetoclax in bone marrow.” The analysis returned a side-by-side differential expression comparison and a pathway enrichment view for each drug, with shared and divergent programs called out.
Results. Cytarabine engaged a broader, deeper set of programs, with strong enrichment in nucleotide metabolism and DNA damage response, consistent with a drug that disrupts DNA synthesis across many cell types. Venetoclax produced a narrower, more focused signature centered on BCL-2 family regulation and apoptotic signaling. The pathway-level view made the divergence between the two drugs immediately visible rather than buried in two gene lists requiring manual cross-reference.
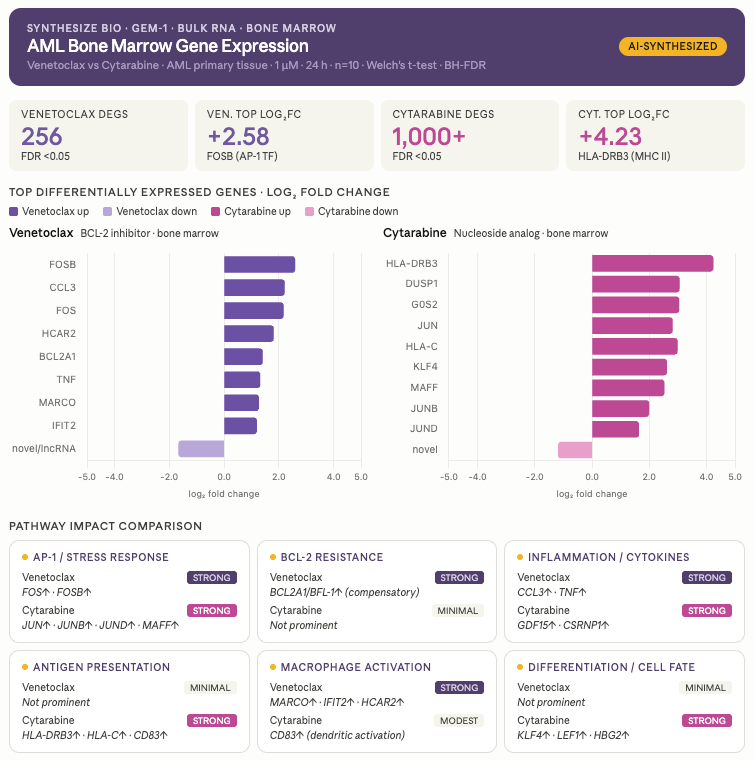
Interpretation. Pathway-level comparison is where biological meaning consolidates, and where most early decisions about mechanism and combination strategy actually get made. Compressing that work from days of setup into a minutes-long conversation meaningfully changes how often it’s worth doing, and therefore how often it gets done.
3. AML cell line selection
Summary. Researchers running experiments on lab-grown cancer cells need to know whether their chosen cell line actually resembles the disease, but that comparison rarely gets done because it’s fiddly to set up. I asked Claude + GEM-1 to rank AML cell lines by similarity to actual AML in bone marrow. After some prompt adjustment (and a workaround for cell lines not represented in the training data), the system delivered ranked candidates with shared and divergent biology called out for each. The value isn’t just the ranking, it’s the explanation of why a line ranks where it does. That’s the difference between a default and a decision.
Details: AML cell line selection
Question. Cell line choice is one of the most consequential early decisions in a preclinical program. It shapes what a study can observe and which signals get missed: if the model doesn’t carry the relevant biology, it can’t return the right measurements. In AML, no single cell line captures the heterogeneity of the disease, and the standard answer — we use what the lab next door uses (no shame, I’ve been there) — can lock programs into the wrong model for the question they’re trying to ask. Grounding the choice in transcriptomic similarity to patient tissue is the right approach, but it rarely gets done because the comparison is difficult to set up. Can Claude + GEM-1 pick the right stand-in cells, and explain its reasoning?
Conversation & context. I asked Claude to use the Synthesize Bio connector to identify which AML-derived cell line has gene expression most similar to actual AML in bone marrow. Some prompt adjustment was needed since Claude didn’t reach for the connector initially. Several of the most commonly used AML lines also weren’t represented in the training data. Once I asked it to find five lines that were represented and rank them, the comparison worked well.
Results. Each line was scored for similarity to primary AML, with shared and divergent biological programs called out alongside the ranking.
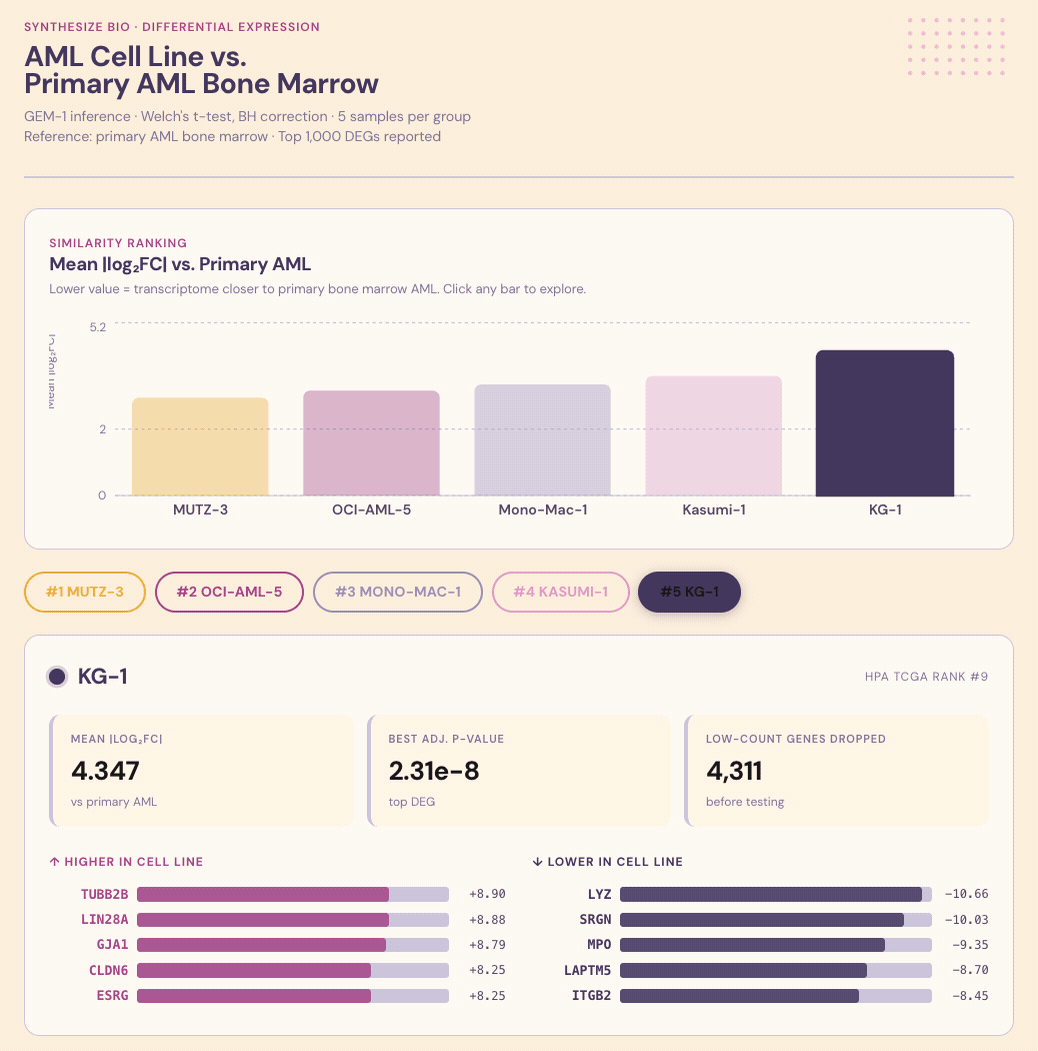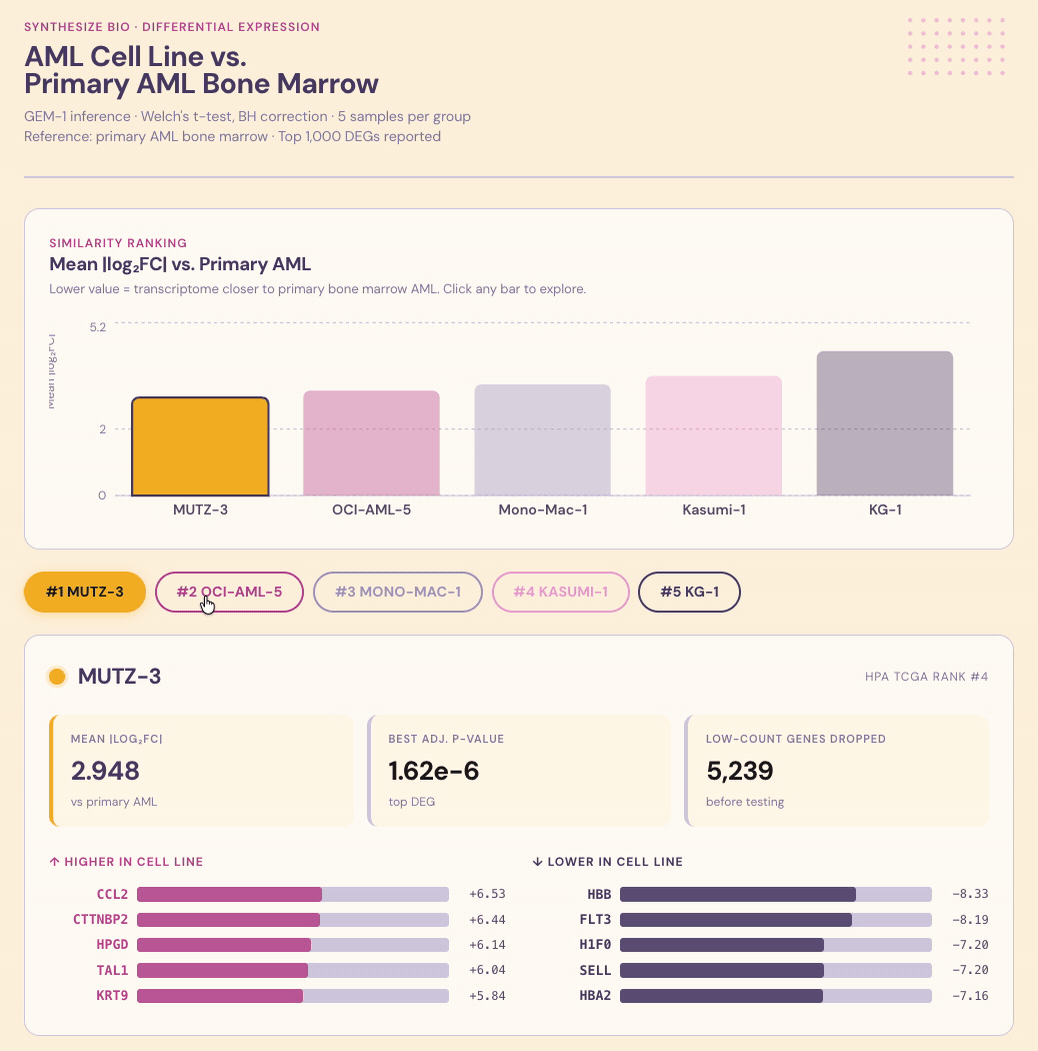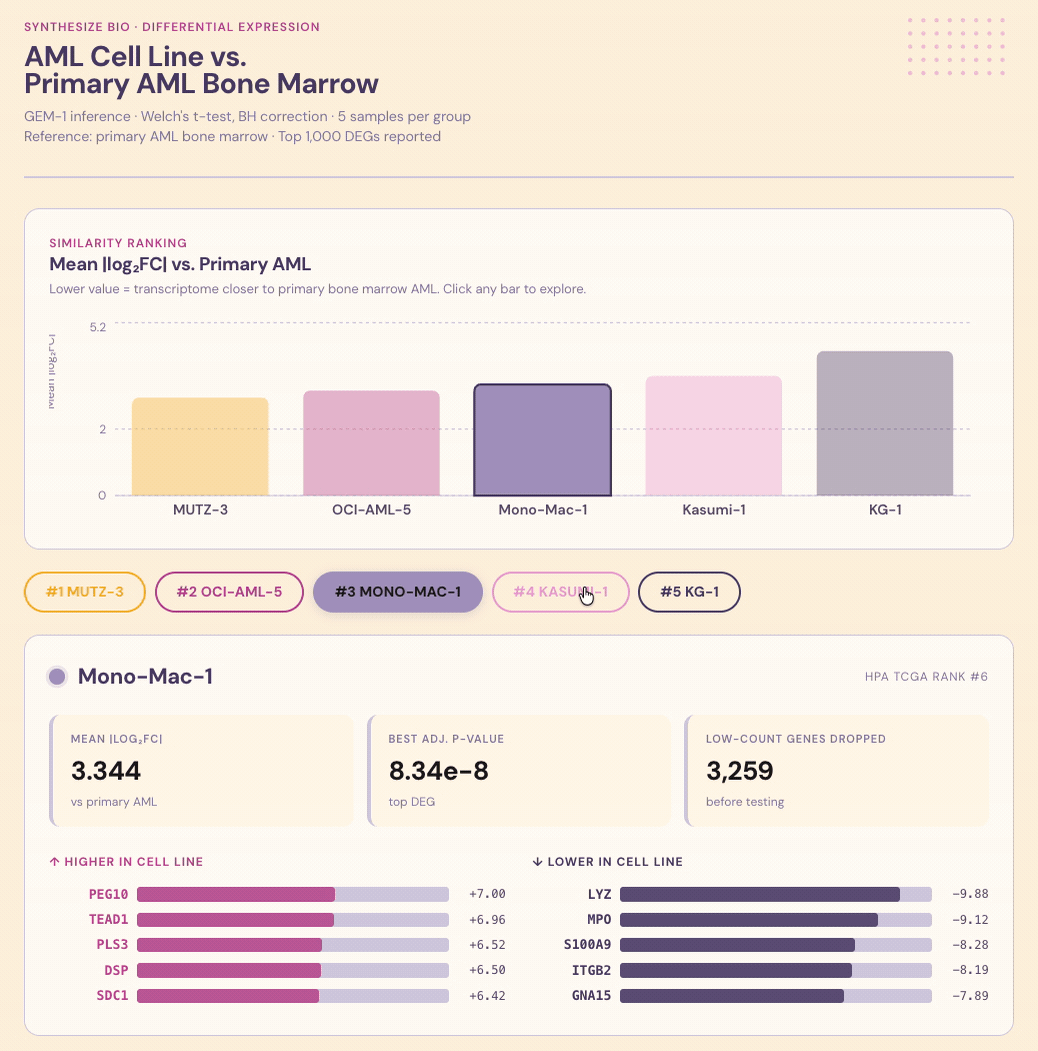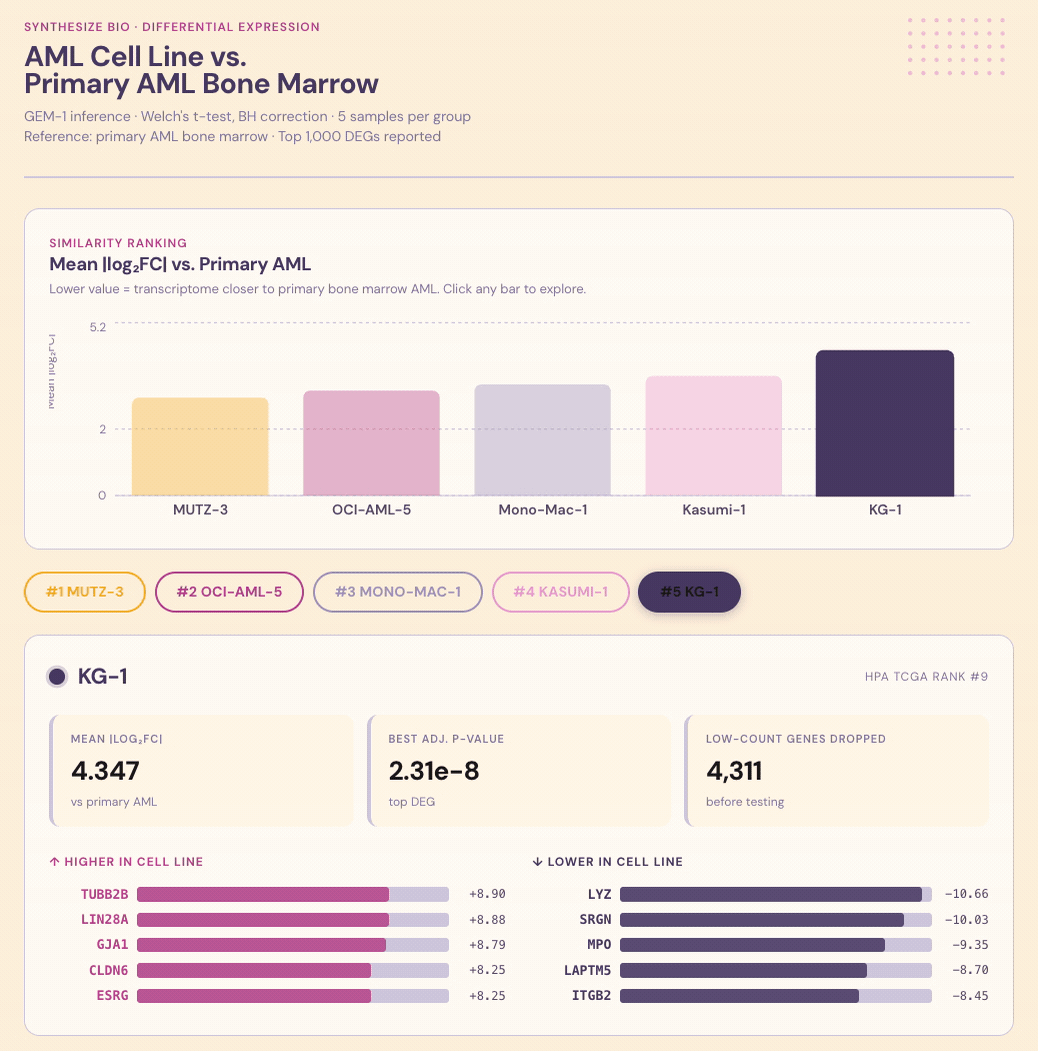
Interpretation. The value isn’t just the ranking, it’s knowing why a line ranks where it does. Which programs it captures faithfully, where it diverges, and whether those divergences matter for the biology you actually care about studying. That’s the kind of context that turns a cell-line choice from a default into a decision that sharpens results and decisions.
Our pharma veteran’s conclusion?
This is the kind of fast-feedback system that I wish I’d had access to during early target validation conversations on dozens of programs. After twenty-plus years on the pharma side of these conversations, that’s not a claim I make casually.
What I found was more grounded than I expected: a powerful system that mostly knows what it can and can’t do, that produces interpretable outputs, and that extends to finding biological trends and placing them in context.
The Synthesize Bio + Claude platform rewards a real hypothesis
The platform compresses weeks of work into minutes for a specific class of questions: whether a perturbation does what you’d expect, whether a disease context adds signal, whether a cell line is a reasonable model for the biology you care about. That’s the stage where it’s most useful: when you have a real biological hypothesis specific enough to test but early enough that a full bioinformatics request isn’t yet justified. The clearer the question going in, the more the tool delivers.
Pathway-level interpretation is consistently where the most useful results appear. Individual gene lists from differential expression are noisy and require deep contextual knowledge to read; pathway enrichment is where biological meaning consolidates. The platform handles this well, and the visualizations are clear enough to share in an internal discussion and to guide decisions.
The conversational format in Claude carries a less obvious benefit: it keeps the iteration loop short. Ask a question, see something unexpected, ask the follow-up — all without rebuilding an analysis from scratch. For exploratory work, that responsiveness matters more than it gets credit for.
Limitations worth knowing
Coverage. Many clinically relevant compounds — particularly those in active development — aren’t represented in the open training data. In every example above, we used a proxy for the therapeutic we actually cared about. That’s not always a problem; mechanistically adjacent compounds can tell you something useful, and the model doesn’t confabulate: when it lacks good data for a drug or cell line, it says so rather than generating plausible-sounding but unreliable predictions. It’s worth being deliberate, though, about whether the proxy is close enough to the compound you actually care about. This coverage limitation applies only to the free, open version. Synthesize Bio can generate data for novel compounds not in the open training corpus — that’s the work we do with our commercial partners, and we’re actively expanding it.
Reporting cap. To keep responses fast and readable in chat, the Synthesize Bio + Claude interface currently surfaces the top 1,000 genes whose activity changed clearly enough to count as a real signal. Subtler shifts live in the underlying Synthesize Bio analysis but aren’t yet displayed in the chat output. Expanding access is on the near-term roadmap.
Framing. The platform answers the question you asked, but it doesn’t push back on how you’ve framed it. With domain expertise this is straightforward; when working adjacent to your primary training, build in a deliberate check on whether the experiment you’ve set up actually maps onto the biology you mean to test. Smarter framing checks are an area of active model development.
For practical tips on getting the most out of the connector — including searching the catalog before launching an experiment and selecting good comparator compounds — see the Synthesize Bio documentation.
From a chat window to a drug program
This is what we believe AI in drug development is going to look like: a virtual scientist working alongside a virtual human laboratory, both serving the human scientists and decision makers asking the questions.
Claude is the virtual scientist — it interprets a question, reasons about what experiments would answer it, connects results to the broader scientific literature, and synthesizes findings into something a researcher can act on. Synthesize Bio’s GEM-1 is the virtual human laboratory — it runs the experiments, generates gene expression data at a depth and breadth no individual wet lab can match. Each is powerful on its own. Together, they let a pharma scientist do something new: test thousands of ideas virtually, contextualize the results against everything that’s already known, and prioritize resources toward the questions most likely to pay off.
The connector in Claude is the earliest expression of that pairing. It’s an entry point that lets individual scientists use Synthesize Bio’s virtual human modeling on their own. The free version is most valuable early in a program, when the questions sound like: does this target behave as expected in the relevant tissue? Is the cell line we’ve been using a reasonable model? In most organizations, exploring the entire hypothesis space is often infeasible, so teams have to choose between breadth and depth." Our virtual human models help teams explore the complex biological space at speed.
What this changes is the economics of a scientific question. Before anyone’s committed, a scientist can run a biological experiment in natural language, get a pathway-level answer in minutes against virtual human tissue data that would otherwise be inaccessible, and have it interpreted against the broader literature in the same conversation. That doesn’t replace scientific judgment. It hands more of it back to the scientist, earlier — when ideas are less expensive to test and decisions are still reversible.
Build the architecture with us
Synthesize Bio is building virtual human models across the drug-development pipeline, with our deepest engagements aimed at clinical trial success in partnership with pharma. The connector is the entry point; the model architecture is much larger and more powerful when customized. Drug developers that adopt this pairing first — virtual scientist plus virtual human laboratory — will move faster, test more ideas, and stop funding the wrong experiments and run the ones that matter most.
If that’s the kind of acceleration you’re trying to build, we should talk.
Key terms used in this post
Virtual human modeling: A virtual human is a computational system designed to simulate human responses under real experimentation. Synthesize Bio's approach combines generative models of human gene expression with downstream bridge models that predict specific human responses.
Transcriptomics: Measuring which genes are active in a tissue.
Differential expression: Which genes changed between conditions.
Pathway enrichment: Which biological programs those genes belong to.
GEM-1: Synthesize Bio’s gene expression foundation model.
Perturbation: An experimental intervention (drug, gene silencing, etc.).
Cell line: Lab-grown cells used as a simple disease model.