
Inside GEM-1: The training data
Building the dataset that powers the next era of virtual tissues

“What’s it trained on?” That’s the question we get all the time since we released GEM-1 in 2025. GEM-1 is our first foundation model that predicts gene expression. Its capabilities are impressive. It can predict the expression of 44,592 genes. As you can imagine, its training data are impressive, too.
Curating those data was a massive undertaking. Public genomic archives contain vast RNA-seq data but often lack the consistent, high-quality curation required to easily define the relationship between an experiment and its outcome. We often get comparisons with protein structure predictions, an endeavor that benefits from well-defined inputs and highly curated databases like the Protein Data Bank. Transcriptomic data are a different beast, with messier fields and more free-text metadata.
In this post, we dive into what biology is represented in the massive dataset of high-quality experimental metadata linked to gene expression data we built to train our first public model.
If you want to go deeper into technical details on the dataset construction, please refer to our preprint on GEM-1!
The goal: Predict gene expression
Our main goal was to address a key problem in science: getting critical transcriptomic data from experiments and clinical trials takes a long time and sometimes isn’t even possible. We thought – what if you could run gene expression experiments in the time it takes to pipette a 96-well plate?
Our team was intimately familiar with both the massive quantities of public RNA-seq data out there and the many challenges with actually using those data for AI. Armed with that knowledge and our mission, we built GEM-1 to turn the global, public archive of human genomics into a predictive engine.
The gene expression data
Training a deep learning model like GEM-1 requires a massive amount of high-quality data. We wanted to train a model that captured both bulk and single-cell RNA-seq. Unlike the Protein Data Bank, the world’s transcriptomic data are a messy library without consistent curation, where scientists use different names for the same things, like “tissue” or “body site.”
For bulk data, we targeted the Sequence Read Archive (SRA), which houses millions of next-generation sequencing records. Specifically, we focused on building a comprehensive human dataset of short-read bulk RNA-seq data. To achieve this, we developed a highly efficient, customized pipeline to process the raw FASTQ files for over half a million samples on AWS. Remarkably, we kept the processing cost under $0.05 per sample, and our small, dedicated team processed 500,000+ samples in under 4 months time. An immense task! This efficiency isn’t just a flex of how awesome our team is, it’s important scientifically. It allowed GEM-1 to see the enormous diversity of human biology represented in the SRA – more than any other model.
For single-cell data, we turned to the CellxGene and scPerturb databases. We used CellxGene for primary tissue data (excludes cell lines and drug screens) and scPerturb for targeted perturbations, which consists mostly of cell lines. Specifically, we incorporated 41,704,552 cells marked as primary data in the July 2024 release of CellxGene, along with 10 datasets from scPerturb. We restricted scPerturb to only a handful of datasets in our first version because modeling perturb-seq data was not a goal.
The sample metadata: Cleaning for clarity
Obtaining and processing gene expression data was the easy part! We also needed clean and accurate descriptions of each experiment and sample. To train a deep learning model like GEM-1 effectively, metadata fields must be consistent. To manage consistency, we restricted each field to a small allowed vocabulary (in technical terms, we mapped to a standardized ontology).
For single-cell data, this was relatively straightforward. CellxGene adheres to a strict schema using ontology terms, requiring minimal conversion. The scPerturb database required slightly more work to map ontology terms but was largely usable intact.
The bulk RNA-seq sample metadata, however, proved much harder. SRA metadata is often unstructured and includes columns named at the depositor’s discretion. That “discretion” leads to wildly different data labels. For example, we needed to know the tissue of origin of samples. Sometimes we found that information in fields labeled anything from “tissue” to “sampling site” to “body site” to “tisue” ([sic] typo!), but most of the time, samples lacked a tissue column entirely. In those cases, we tried to extract the information from the abstract or sample identifier.
We thought, why not use AI to build AI? We did! To tackle the wild west problem above, we built a metadata agent using LLMs and defined heuristics to extract and structure metadata and map fields to standard ontologies.
The harmonized labels: clean metadata
The metadata fields were categorized into three groups - biological, technical, and perturbational - which correspond to the three latent spaces learned by GEM-1.
Metadata Fields:
Biological: age, cell line, cell type, disease, sample type (e.g. primary, cell line, xenograft), sex, and tissue.
Perturbational: perturbation, dose, time, and type.
Technical: Varies between bulk and single-cell data.
Bulk Data: Library selection method, library layout (single or paired end), and instrument.
Single-Cell Data: Assay.
This difference in technical fields is due in part to the distinct data types and the databases utilized. Notably, the SRA and CellxGene databases provide these technical fields with 100% labeling for all samples.
These three groups enabled our model to differentiate between technical effects (which, although important, usually aren’t what we ultimately care about as biologists) and real biological phenomena.
Sample types, sex and age
As noted earlier, CellxGene consists largely of primary samples, while scPerturb is dominated by cell lines. Our bulk expression data offers a balance, split between primary samples (~51%) and cell lines (~45%), with a small fraction (~4%) categorized as “other” (including organoids and xenografts).
The sex ratio within each bulk sample type is generally balanced, though nearly half of the primary and “other” samples lack sex labels in the metadata. Cell lines have a much higher labeling rate because we can infer sex directly from the cell line identity. In the CellxGene data (top right), we observe a slight bias toward male cells.
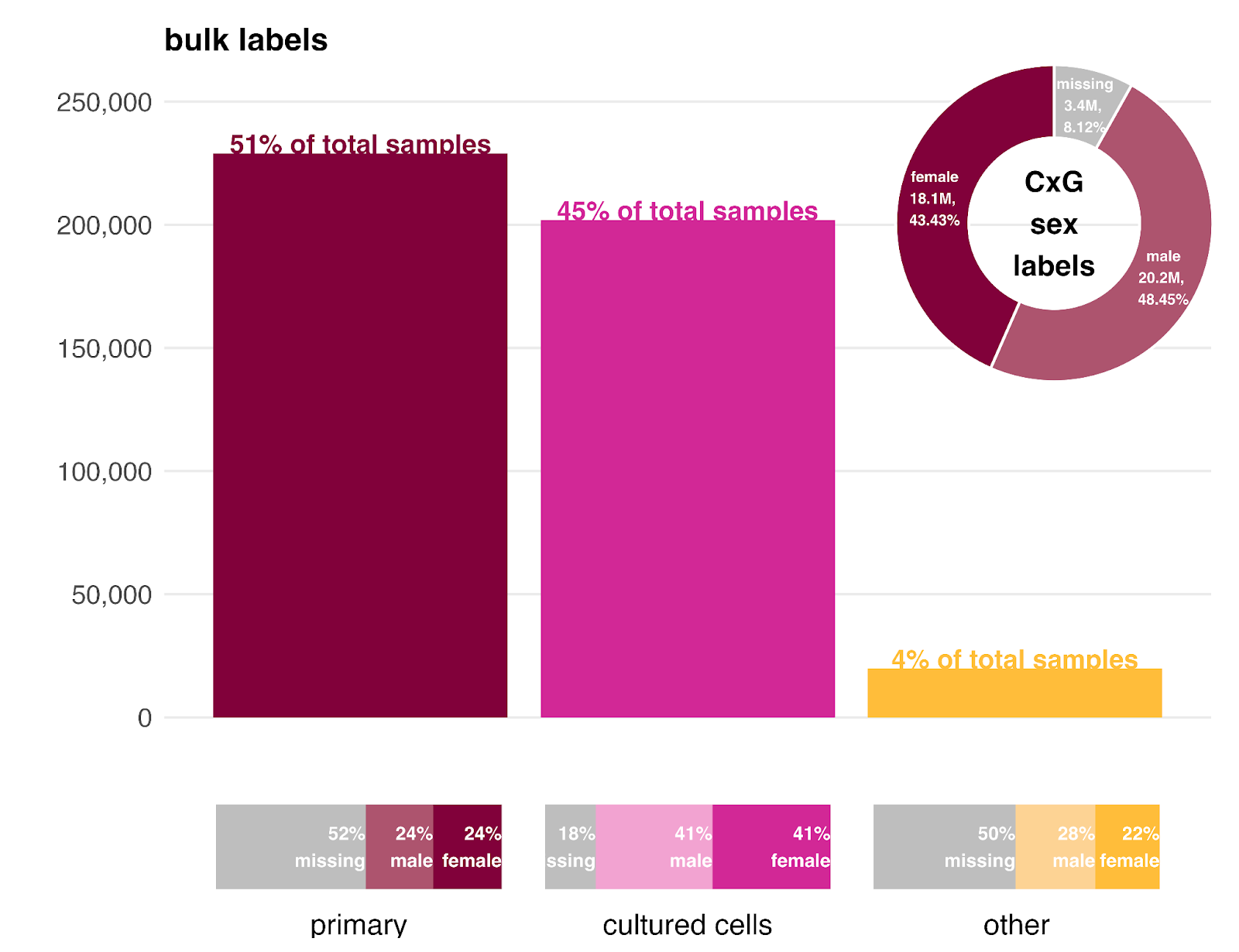
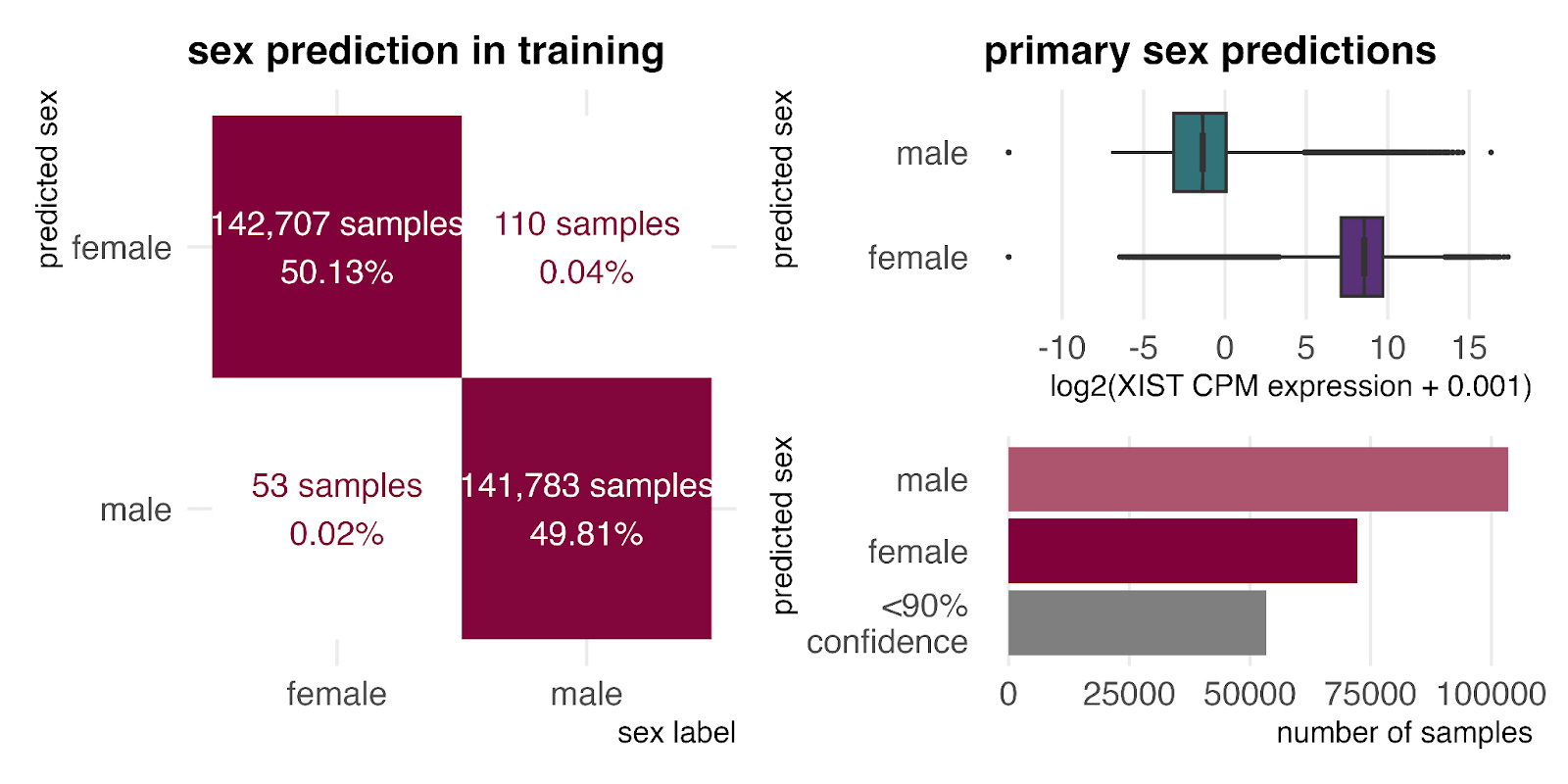
One advantage of GEM-1 is that it learns to predict missing metadata during training. We used this capability to estimate the sex ratio for the unlabeled primary samples.
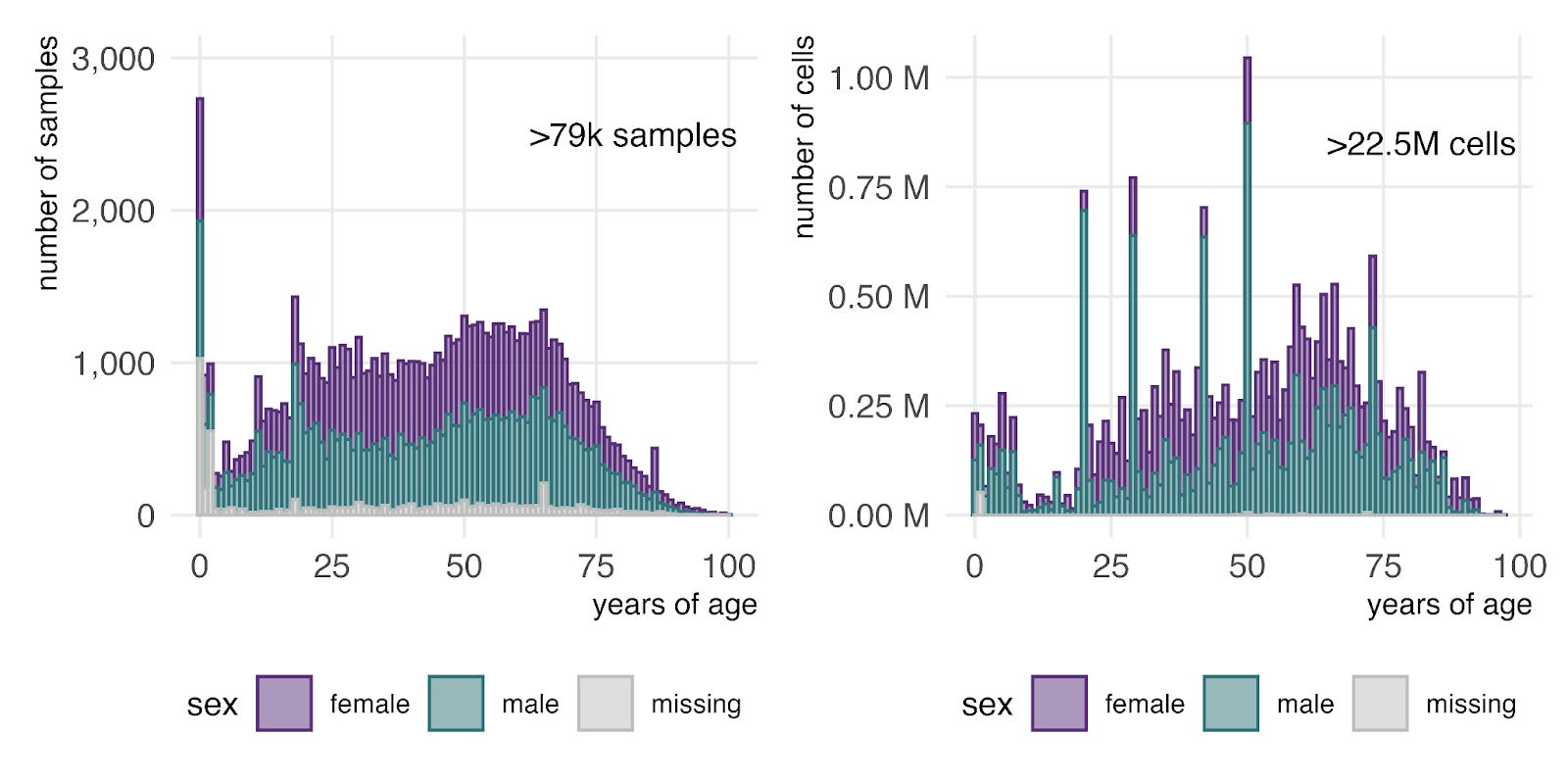
Finally, examining age distributions in primary bulk RNA-seq (left) and CellxGene single-cell RNA-seq (right) shows that the entire human lifespan is represented in both datasets. As expected, the data is weighted toward middle age, with fewer samples representing the very youngest and oldest humans.
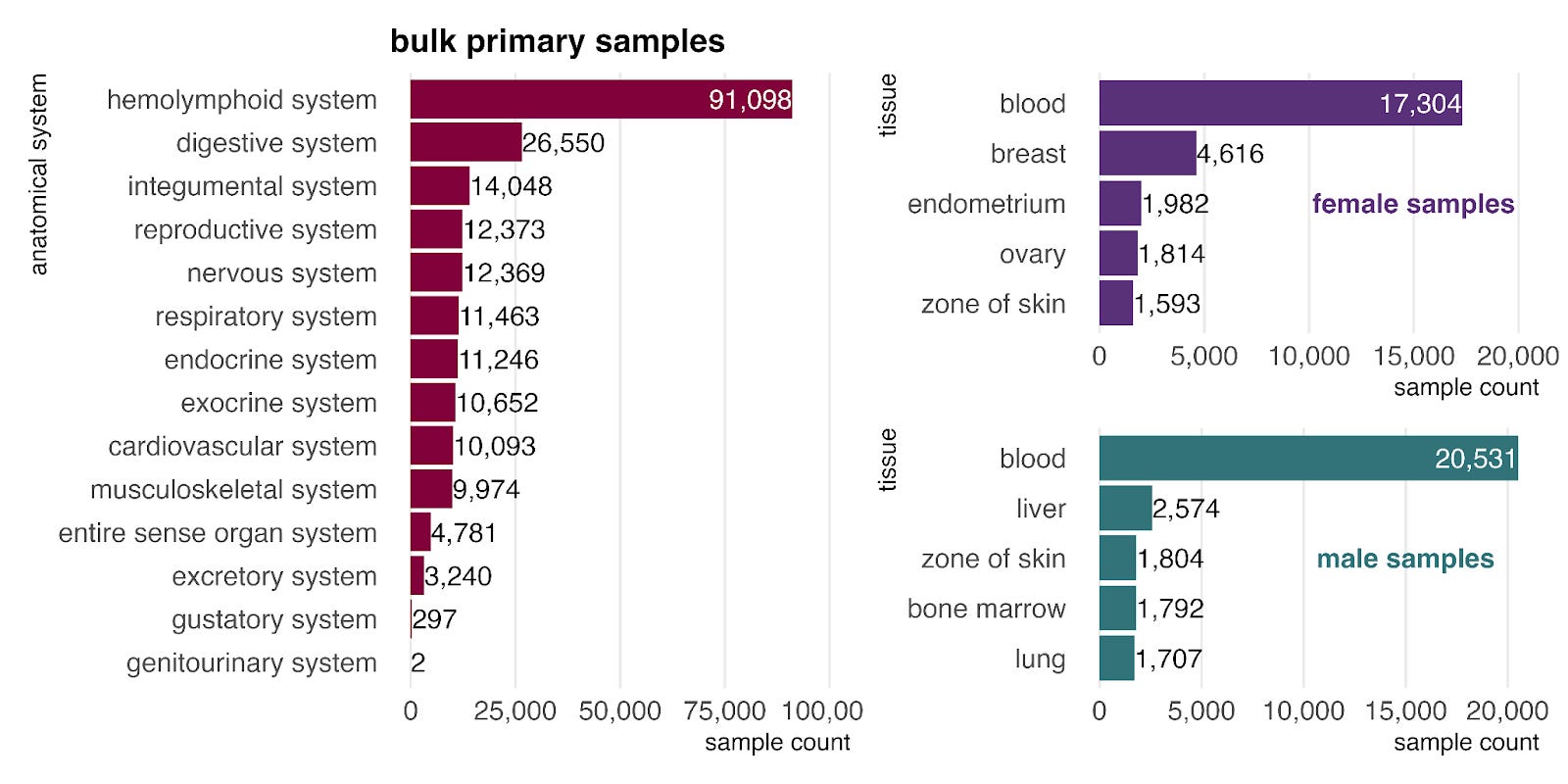
Primary tissue representation
In the bulk RNA-seq data, primary samples are most commonly from the hemolymphoid system (left), mostly blood, reflecting its high accessibility for sampling. When stratified by sex (right), blood remains the most common tissue for both groups, and female-labeled samples show a clear bias toward reproductive tissues.
Unlike bulk, the single-cell (CellxGene) data is enriched for a higher proportion of nervous system samples, comprised mostly of various brain tissues. As in bulk, we continue to see a higher prevalence of breast tissue cells in the female dataset.
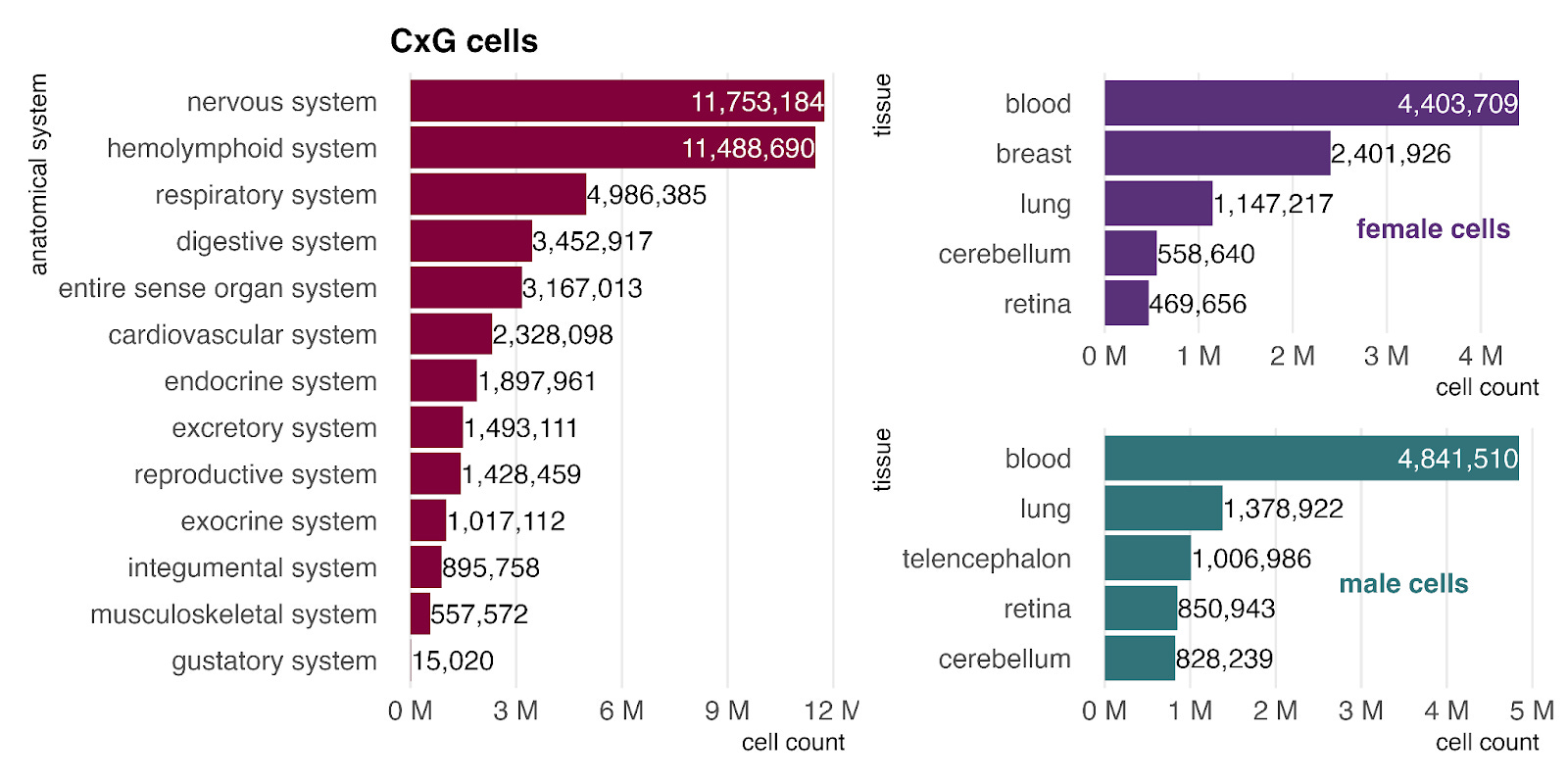
Primary disease representation
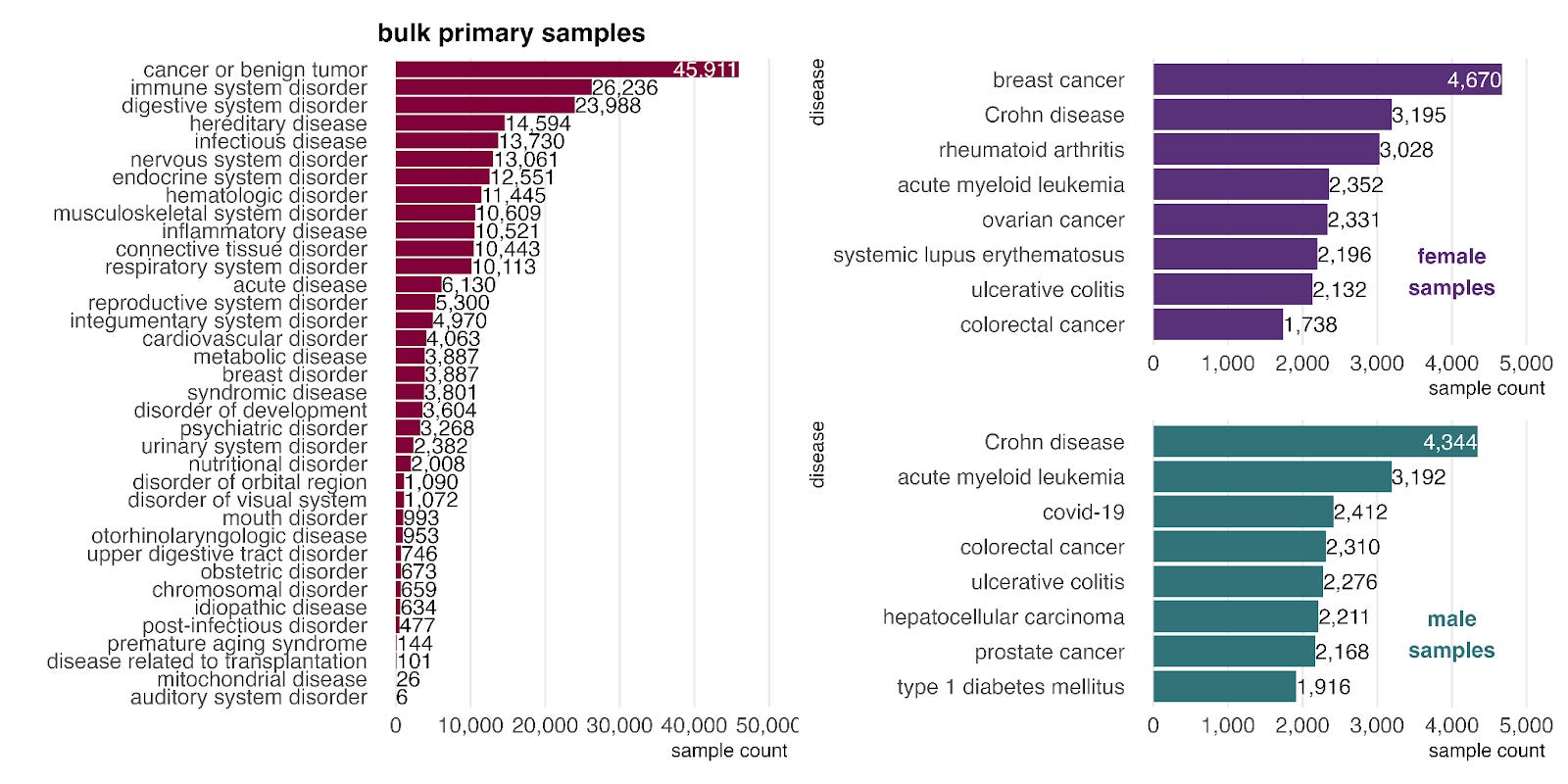
How biased are our training data towards certain diseases and phenotypes? Cancers and digestive disorders, such as Crohn’s disease and ulcerative colitis, are prominent, likely due to the clinical course of care including diagnostic biopsies.
When stratified by sex, cancers and digestive disorders are prominent, likely due to the frequent use of biopsies in these conditions. Beyond these common categories, we observe a few sex-biased patterns. In female samples (top right), autoimmune diseases like rheumatoid arthritis and systemic lupus erythematosus are highly represented, consistent with their known higher prevalence in females (citation). In male samples (bottom right), COVID-19 and type 1 diabetes mellitus appear among the top diseases. This aligns with clinical observations that COVID-19 severity is often greater in males (citation) and that type 1 diabetes shows a male predominance in certain high-risk populations (citation).
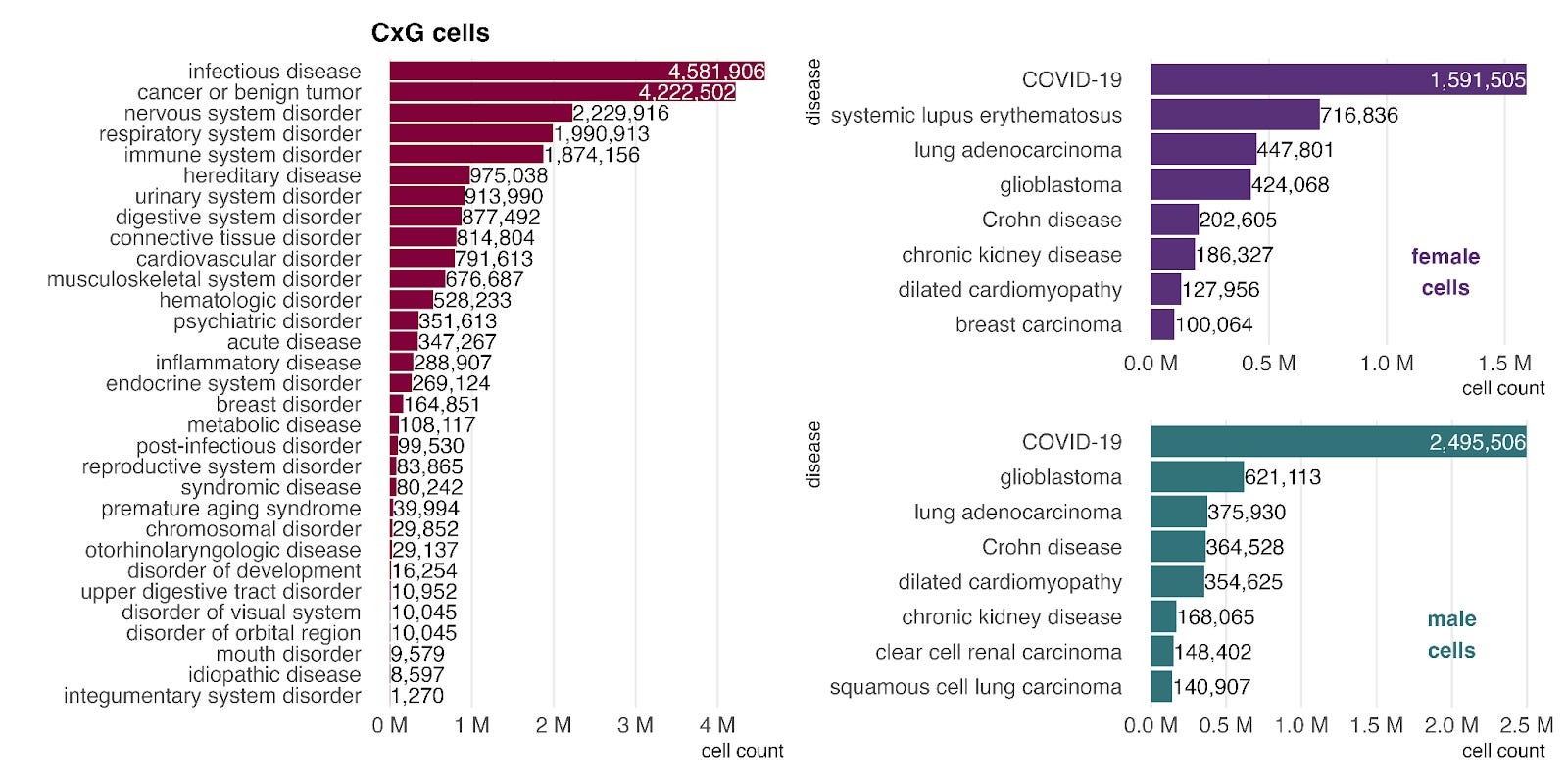
The single-cell data from CellxGene have parallels with the bulk data, particularly in the strong representation of cancer. However, the impact of the COVID-19 pandemic is even more pronounced here. COVID-19 is the most common disease label across both sexes (right) and accounts for over 96% of cells in the infectious disease category, driving its position as the top disease group in CellxGene.
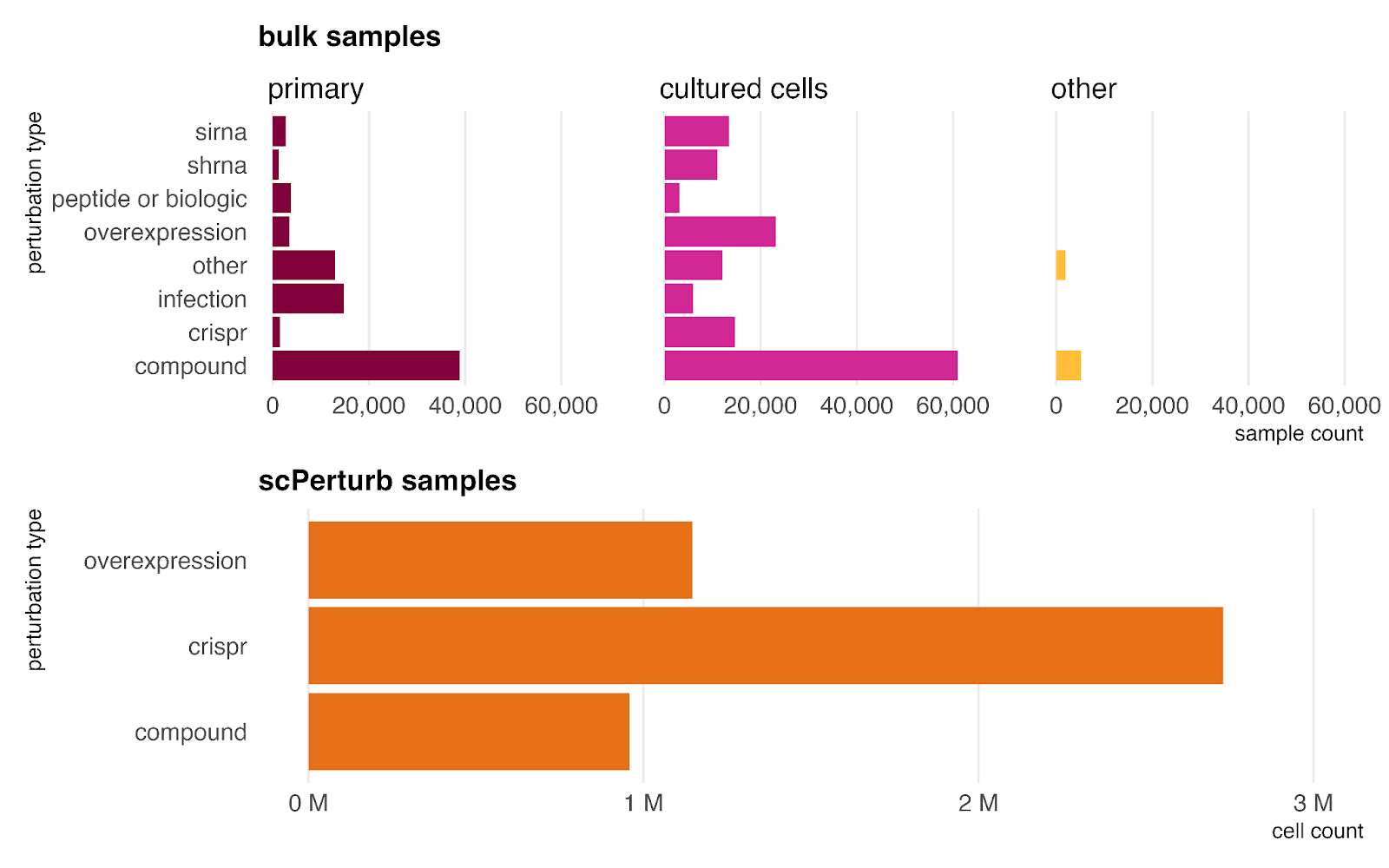
Perturbations
Our dataset incorporates perturbed samples from both bulk RNA-seq and single-cell sources (scPerturb). These are categorized into five key types: genetic perturbations (siRNA, shRNA, CRISPR, overexpression); peptides or biologics; compounds (including drugs and environmental exposures); infections; and other (covering factors not in other categories like diet, hypoxia, and mechanical stress).
Because experimental perturbations are significantly easier to perform in in vitro settings than in human subjects, the majority of these samples (including the entire scPerturb dataset) are derived from cultured cells rather than primary tissues. However, all types are also represented by at least 1,000 samples in primary data, ensuring the model learns to generalize perturbation effects to the primary setting as well.
Looking forward to our next generation of models
Constructing the dataset for GEM-1 provided valuable lessons in extracting metadata from free text, mapping terms to ontologies, and designing effective schemas for model training. Armed with these insights, we have already begun paving the way for the next generation of models. By leveraging more powerful LLMs, refined mapping strategies, and a redesigned perturbation schema, we have significantly increased both the volume of labels and the precision of ontology mapping. We have also added more expression data. These advancements set a strong foundation for GEM-2, promising an even more robust and capable model.